Introduction¶
LaminDB is an open-source data framework for biology:
{include}
:start-line: 6
:end-line: -4
:::{dropdown} LaminDB features
{include}
:::
LaminHub is a data collaboration hub built on LaminDB similar to how GitHub is built on git.
:::{dropdown} LaminHub features
{include}
:::
Basic features of LaminHub are free. Enterprise features hosted in your or our infrastructure are available on a paid plan!
Quickstart¶
{warning}
Public beta: Close to having converged a stable API, but some breaking changes might still occur.
You'll ingest a small dataset while tracking data lineage, and learn how to validate, annotate, query & search.
Setup¶
Install the lamindb Python package:
pip install 'lamindb[jupyter,bionty]'
Initialize a LaminDB instance mounting plug-in {py:mod}bionty for biological types:
import lamindb as ln
# artifacts are stored in a local directory `./lamin-intro`
# ln.setup.init(schema="bionty", storage="./lamin-intro")
# tag your code with auto-generated identifiers for a script or notebook
ln.transform.stem_uid = "FPnfDtJz8qbE"
ln.transform.version = "1"
# track the execution of a transform with a global run context
ln.track()
💡 connected lamindb: laminlabs/lamindata 💡 notebook imports: anndata==0.10.5.post1 bionty==0.41.1 lamindb==0.68.0 pandas==1.5.3 💡 loaded: Transform(uid='FPnfDtJz8qbE5zKv', name='Introduction', short_name='introduction', version='1', type='notebook', updated_at=2024-03-11 16:08:43 UTC, created_by_id=9) 💡 loaded: Run(uid='39aA4QIBAAYKK3OqAWNk', run_at=2024-03-11 18:39:32 UTC, transform_id=62, created_by_id=9)
Manage artifacts¶
import pandas as pd
# dummy data
df = pd.DataFrame(
{"CD8A": [1, 2, 3], "CD4": [3, 4, 5], "CD14": [5, 6, 7], "perturbation": ["DMSO", "IFNG", "DMSO"]},
index=["observation1", "observation2", "observation3"],
)
With {class}~lamindb.Artifact, you can manage data batches & models in storage as files, folders or arrays.
artifact = ln.Artifact.from_df(df, description="my RNA-seq", version="1")
❗ returning existing artifact with same hash: Artifact(uid='S7YB8PFudvFudzXzQMRy', suffix='.parquet', accessor='DataFrame', description='my RNA-seq', version='1', size=4122, hash='iRFVECWdNmTqDesUtIE37A', hash_type='md5', visibility=1, key_is_virtual=True, updated_at=2024-03-11 18:33:23 UTC, storage_id=2, transform_id=62, run_id=77, created_by_id=9)
Any artifact comes with typed, relational metadata:
artifact.describe()
Artifact(uid='S7YB8PFudvFudzXzQMRy', suffix='.parquet', accessor='DataFrame', description='my RNA-seq', version='1', size=4122, hash='iRFVECWdNmTqDesUtIE37A', hash_type='md5', visibility=1, key_is_virtual=True, updated_at=2024-03-11 18:33:23 UTC) Provenance: 🗃️ storage: Storage(uid='D9BilDV2', root='s3://lamindata', type='s3', region='us-east-1', updated_at=2023-12-07 09:50:27 UTC, created_by_id=2) 📔 transform: Transform(uid='FPnfDtJz8qbE5zKv', name='Introduction', short_name='introduction', version='1', type='notebook', updated_at=2024-03-11 16:08:43 UTC, created_by_id=9) 👣 run: Run(uid='39aA4QIBAAYKK3OqAWNk', run_at=2024-03-11 18:39:32 UTC, transform_id=62, created_by_id=9) 👤 created_by: User(uid='FBa7SHjn', handle='falexwolf', name='Alex Wolf', updated_at=2023-10-19 18:58:28 UTC) Features: columns: FeatureSet(uid='5o3wKfvHbv7sXzFDuyH4', n=4, registry='core.Feature', hash='3M6xerbxJQKIH0SvRy9L', updated_at=2024-03-11 18:33:37 UTC, created_by_id=9) CD8A (number) CD4 (number) CD14 (number) 🔗 perturbation (2, core.ULabel): 'DMSO', 'IFNG' Labels: 🏷️ ulabels (3, core.ULabel): 'DMSO', 'IFNG', 'Candidate marker study'
If you save an artifact, you'll save data & metadata in one operation:
artifact.save()
For any artifact, you can view its data lineage:
artifact.view_lineage()

:::{dropdown} Data provenance in the UI
The screenshot shows a notebook with its latest report, runs, output files, and parent notebooks. On the run view, you'll see input files.
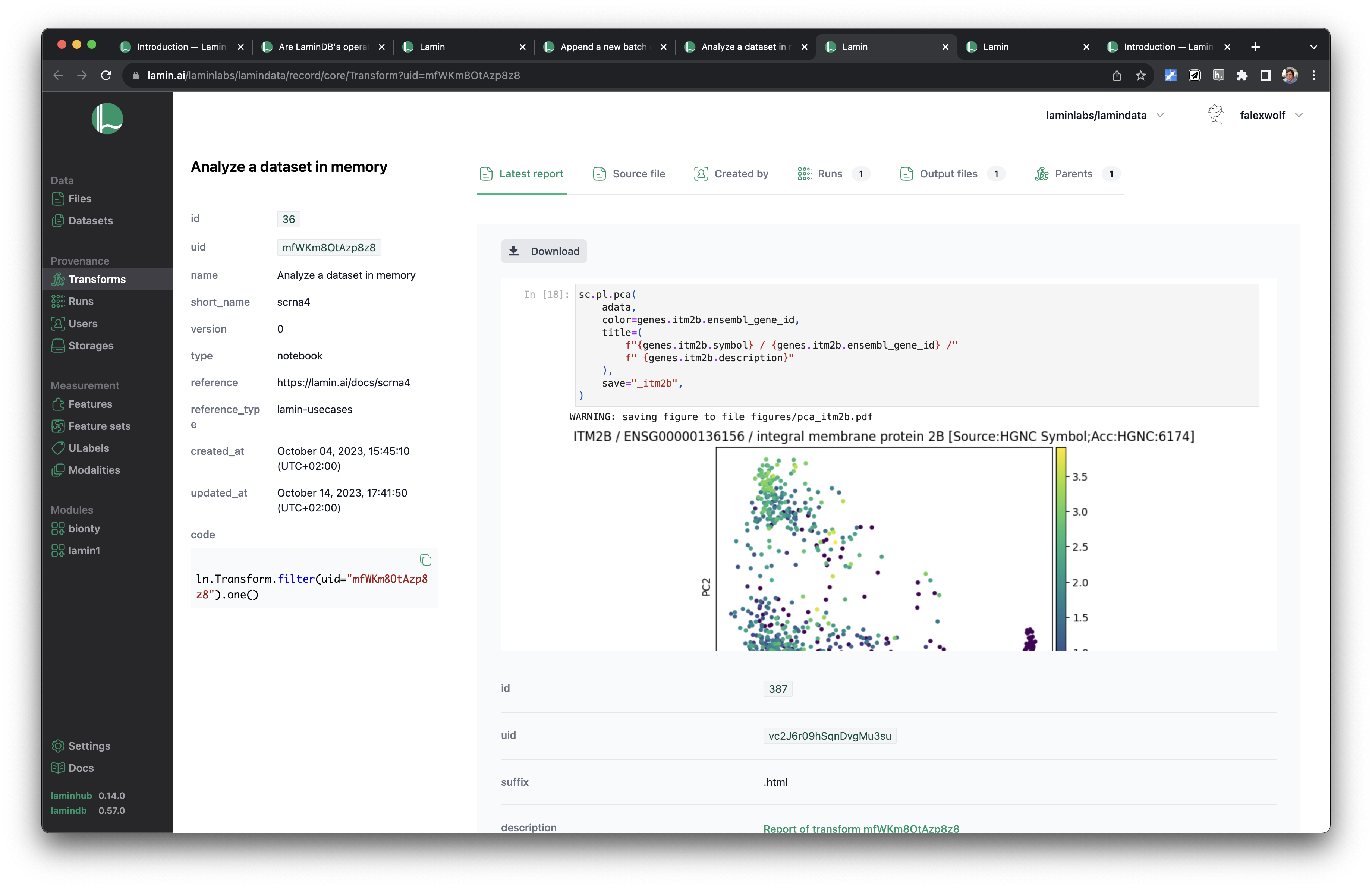
:::
Loading an artifact returns an object determined by its .accessor and .suffix:
artifact.load()
| CD8A | CD4 | CD14 | perturbation | |
|---|---|---|---|---|
| observation1 | 1 | 3 | 5 | DMSO |
| observation2 | 2 | 4 | 6 | IFNG |
| observation3 | 3 | 5 | 7 | DMSO |
Query¶
A simple query:
ln.Artifact.filter(description="my RNA-seq").df()
| uid | storage_id | key | suffix | accessor | description | version | size | hash | hash_type | n_objects | n_observations | transform_id | run_id | visibility | key_is_virtual | created_at | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||
| 466 | S7YB8PFudvFudzXzQMRy | 2 | None | .parquet | DataFrame | my RNA-seq | 1 | 4122 | iRFVECWdNmTqDesUtIE37A | md5 | None | None | 62 | 77 | 1 | True | 2024-03-11 16:09:03.373561+00:00 | 2024-03-11 18:39:39.936754+00:00 | 9 |
To query all artifacts ingested from a notebook with title "Introduction":
ln.Transform.filter(name="Introduction").df()
| uid | name | short_name | version | type | latest_report_id | source_code_id | reference | reference_type | created_at | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||
| 53 | FPnfDtJz8qbEz8 | Introduction | introduction | 0 | notebook | 445.0 | 443.0 | https://lamin.ai/docs/introduction | lamindb guide | 2023-11-03 14:29:45.251914+00:00 | 2023-12-11 10:18:42.624596+00:00 | 2 |
| 62 | FPnfDtJz8qbE5zKv | Introduction | introduction | 1 | notebook | NaN | NaN | None | None | 2024-03-11 16:08:43.875316+00:00 | 2024-03-11 16:08:43.875330+00:00 | 9 |
transform = ln.Transform.filter(name="Introduction", version="1").one()
artifacts = ln.Artifact.filter(transform=transform).all()
Because, under-the-hood, LaminDB is SQL & Django, you can write arbitrarily complex relational queries:
artifacts = ln.Artifact.filter(transform__name__icontains="intro", created_by__handle="anonymous").all()
:::{dropdown} Query in the UI
If you work with a remote instance on LaminHub, you can compose queries as shown below.
Because LaminDB's metadata-management is based on SQL, registries can easily have 10s of millions of rows.
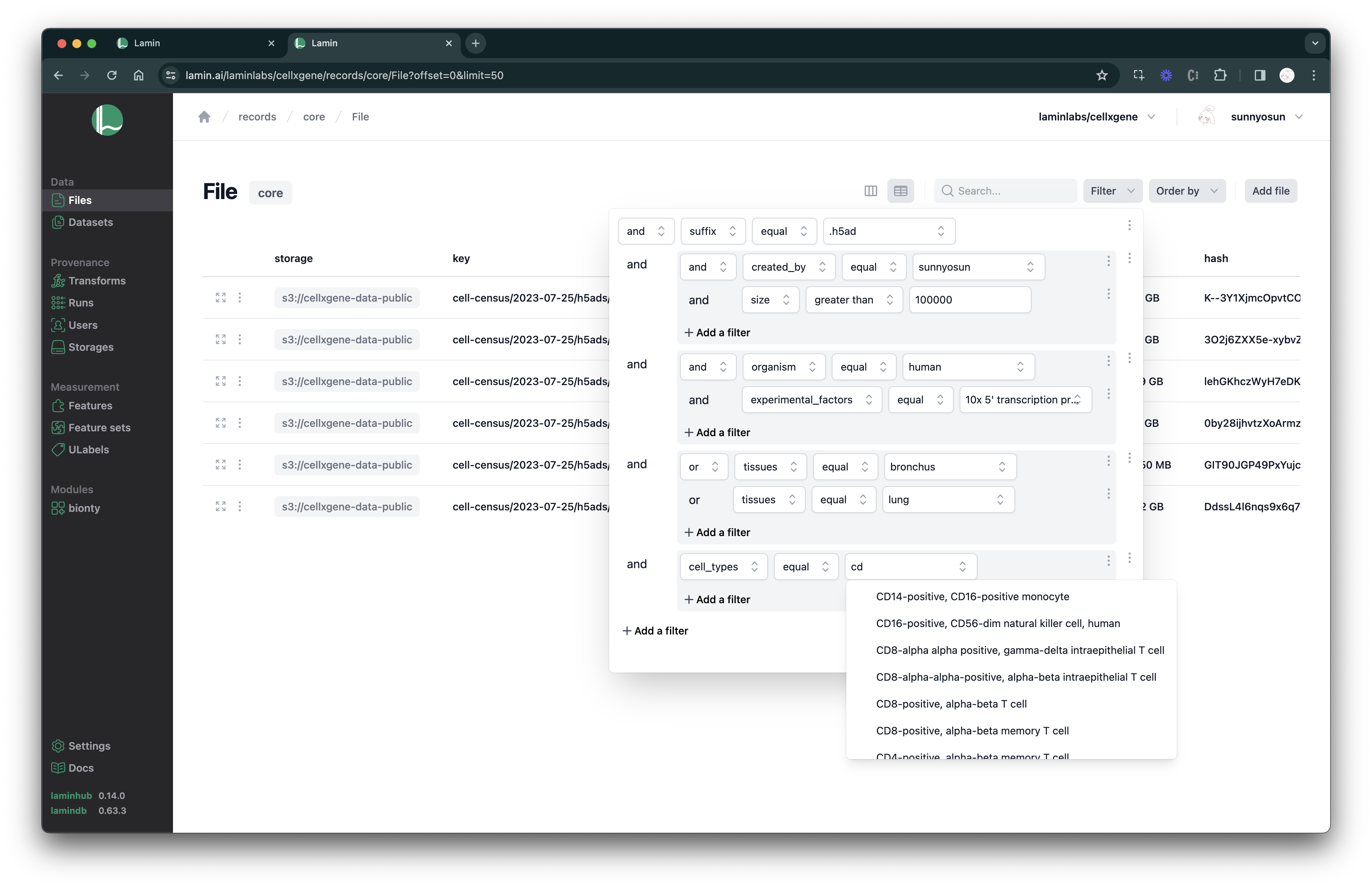
:::
Search¶
Search the {class}~lamindb.Artifact registry:
ln.Artifact.search("RNAseq")
| key | description | score | |
|---|---|---|---|
| uid | |||
| YdTtu9TBPYe6ov1ItOVK | scRNA-seq pbmc68k reduced | 75.0 | |
| myPxexSQMW3Xrdz5C31U | scRNA-seq pbmc3k processed | 75.0 | |
| S7YB8PFudvFudzXzQMRy | my RNA-seq | 75.0 | |
| SAPKv9zC4hBmXsHvdReg | scRNA-seq mouse lymph_node | 75.0 | |
| QljH2b4vYYCJmN0kIrc5 | analyzed data of perturbseq | 65.5 | |
| wQdAoegRqgY8p0U8D7qV | perturbseq counts | 62.2 | |
| QljH2b4vYYCJmN0kIrc5 | perturbseq_analyzed.h5ad | 62.2 | |
| hF4cCB0ShpGifVr4kl1O | fastq/schmidt22_perturbseq_R2_001.fastq.gz | 60.0 | |
| 3q7AhNt3DId2KoCdS206 | requirements.txt | 60.0 | |
| oNOA2Ham9HQP1NDMxds7 | requirements.txt | 60.0 | |
| vCt2evuKcBuE6JsdAxrW | requirements.txt | 60.0 | |
| uAAHUIGb13zvUYkMRsGR | CRISPRa Perturb-seq of primary human T cells | 60.0 | |
| wQdAoegRqgY8p0U8D7qV | schmidt22_perturbseq.h5ad | 60.0 | |
| wYYqsieWJfrfT32FHocQ | fastq/schmidt22_perturbseq_R1_001.fastq.gz | 60.0 | |
| TnGJD1EUywW4ehXcZLRC | rna-seq-results/multiqc/star_salmon/multiqc_da... | 60.0 | |
| Mn0Hnty6eKQqNxQDZW6D | rna-seq-results/multiqc/star_salmon/multiqc_da... | 60.0 | |
| xJkeL0OxEFIpvGWKdpne | scrna/conde22.h5ad | 57.0 | |
| wVCsDZhRdZyR3DoNwZ7r | rna-seq-results/multiqc/star_salmon/multiqc_pl... | 50.0 | |
| OxAHARtY0vkGzyKnBZN8 | rna-seq-results/multiqc/star_salmon/multiqc_da... | 50.0 | |
| KTc7MpZ1uDzzqGiIYPQi | rna-seq-results/multiqc/star_salmon/multiqc_da... | 50.0 | |
| qvX9D4mmcS42lie6NQ6V | rna-seq-results/multiqc/star_salmon/multiqc_da... | 50.0 | |
| sBRctkYkXN1mVRiHd4v5 | rna-seq-results/multiqc/star_salmon/multiqc_pl... | 50.0 | |
| HYQ5fpERtBTUk50sFjNb | rna-seq-results/multiqc/star_salmon/multiqc_pl... | 50.0 | |
| xRhXxrlWnX9zq8wo1BOE | rna-seq-results/multiqc/star_salmon/multiqc_pl... | 50.0 | |
| fPKHLwd1P23elLThiQoe | rna-seq-results/multiqc/star_salmon/multiqc_pl... | 50.0 | |
| dfBnbGJEWKjxjvwFugWi | rna-seq-results/multiqc/star_salmon/multiqc_pl... | 50.0 | |
| RuV0HbcrDaQ1J0QKQA3B | rna-seq-results/multiqc/star_salmon/multiqc_pl... | 50.0 | |
| 4woYfGFfN7lbdW6Yph5e | rna-seq-results/multiqc/star_salmon/multiqc_da... | 50.0 | |
| tRislc7bzveWmMdalBBT | rna-seq-results/multiqc/star_salmon/multiqc_da... | 50.0 | |
| U7JZLhQBbjfZJJbAaq1t | rna-seq-results/multiqc/star_salmon/multiqc_pl... | 50.0 | |
| fhtNpEq5EJmI4Dalp0pN | See dataset fhtNpEq5EJmI4Dalp0pN | 46.6 | |
| ckBmWq8Bqzmej574ogH5 | 10x reference adata | 46.6 | |
| Ff4XZtm6Lko0Fz3ysIEl | 10x reference adata | 46.6 | |
| 6fBzkHfG2f0ikIHn415P | Source of transform Qr1kIHvK506r5zKv | 45.0 | |
| t9H3W4sQDpitGJ94Bt31 | Source of transform ManDYgmftZ8C5zKv | 45.0 | |
| j4aAAsQlS4Ud9xbARoAF | Source of transform FPnfDtJz8qbEz8 | 45.0 | |
| kReqQ8BoaXlvWyzeAsiS | Report of transform Nv48yAceNSh85zKv | 45.0 | |
| gcpSpg8NwHvciMandEdP | Report of transform ManDYgmftZ8C5zKv | 45.0 | |
| f2O2dRwuUdy9lOhfq0bF | Report of transform FPnfDtJz8qbEz8 | 45.0 | |
| 3JgPDL5l9kTJUDHkLMd2 | Report of transform FPnfDtJz8qbEz8 | 45.0 | |
| MbTyShN2FrU9IHKDiMwx | Source of transform Nv48yAceNSh85zKv | 45.0 |
Or search any other registry, e.g., {class}~lamindb.Transform:
ln.Transform.search("intro")
| uid | score | |
|---|---|---|
| name | ||
| Introduction | FPnfDtJz8qbEz8 | 90.0 |
| Introduction | FPnfDtJz8qbE5zKv | 90.0 |
| Integrate lung datasets | 0poBAqTX7A0hUb | 67.5 |
| Query & integrate data | wukchS8V976Uz8 | 54.0 |
| Manage a cell type registry | s5mkN5NQ1ttIwy | 54.0 |
| Train an ML model on a collection | Qr1kIHvK506r5zKv | 51.4 |
| Tracking Bulk RNA-seq Nextflow runs | 8124Vtle6ZrOz8 | 51.4 |
| Ingest Tabula Sapiens Lung | 6wW30APjv7kJif | 51.4 |
| .fcs file ingestion | z59qmjSEN1Hn6l | 45.0 |
| Flow cytometry | OWuTtS4SAponz8 | 45.0 |
| Schmidt 2022 | 73HHY1v2yZJfmL | 42.8 |
| Tutorial: Features & labels | dMtrt8YMSdl6z8 | 40.0 |
| Iteratively train an ML model on a dataset | Qr1kIHvK506rz8 | 40.0 |
| Cell Ranger | mpw5kMV0z5G5sM | 40.0 |
| Chromium 10x upload | J5ZTmVxSch3UmX | 40.0 |
| Tutorial: Files & datasets | NJvdsWWbJlZSz8 | 40.0 |
| Preprocess Cell Ranger outputs | piG5scNASXcc0b | 38.0 |
| Query artifacts | agayZTonayqA5zKv | 36.0 |
| Hit identification - genome-wide CRIPSRa IFNG screen of T cells | PtTXoc0RbOIqFn | 36.0 |
| Correlating PDL1 coefficients with Schmidt22 | pdMSzE2X72aey9 | 36.0 |
Look up¶
We can look up records in any registry with auto-complete until we have more than 200k entries:
users = ln.User.lookup()
:::{dropdown} Show me a screenshot
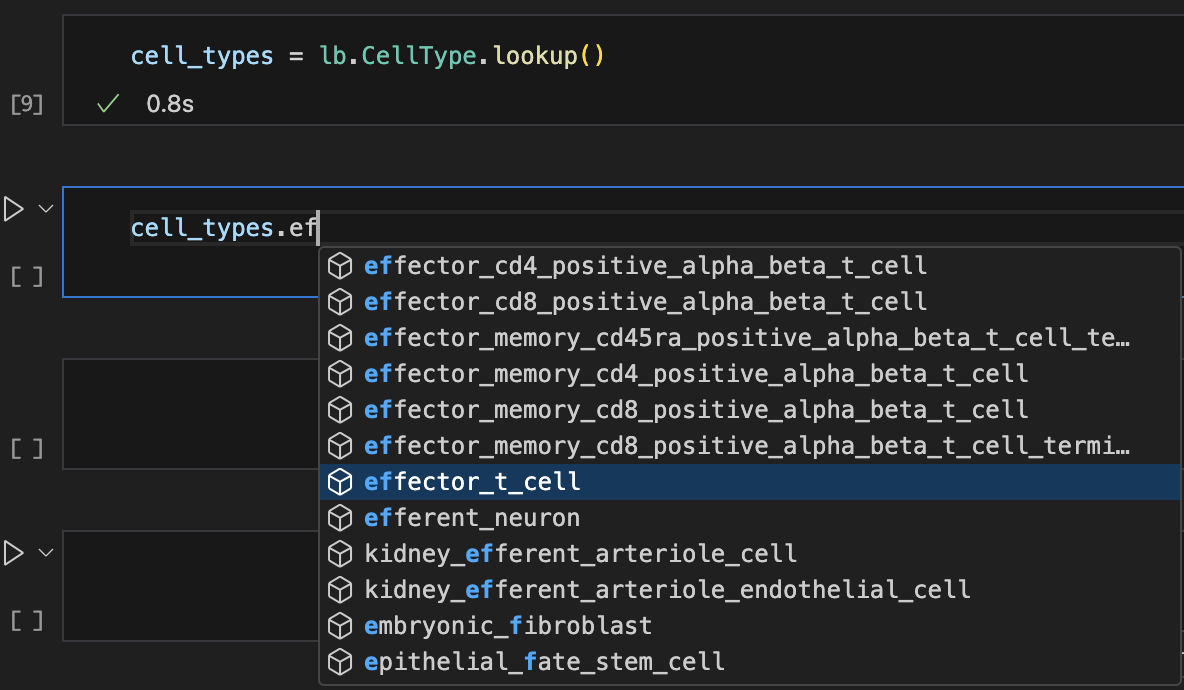
:::
Features & labels¶
For instance, populate the feature registry ({class}~lamindb.Feature) based on the columns measured in the DataFrame:
features = ln.Feature.from_df(df)
ln.save(features)
The registry now looks like this:
ln.Feature.df()
| uid | name | type | unit | description | registries | synonyms | created_at | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||
| 317 | 7nXGhSQJm4Gg | CD14 | number | None | None | None | None | 2024-03-11 16:09:12.552454+00:00 | 2024-03-11 16:09:12.552460+00:00 | 9 |
| 316 | Ep63VlWJffhL | CD4 | number | None | None | None | None | 2024-03-11 16:09:12.552402+00:00 | 2024-03-11 16:09:12.552406+00:00 | 9 |
| 315 | hKqWcJsyRo0x | CD8A | number | None | None | None | None | 2024-03-11 16:09:12.552325+00:00 | 2024-03-11 16:09:12.552338+00:00 | 9 |
| 266 | zvyDVbZln36o | donor | category | None | None | core.ULabel | None | 2023-07-14 19:58:15.852686+00:00 | 2024-01-08 12:27:43.675617+00:00 | 6 |
| 174 | aci5k3PxFx7f | organism | category | None | None | bionty.Organism | None | 2023-07-14 12:41:14.293496+00:00 | 2024-01-08 12:27:43.319385+00:00 | 5 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 5 | b1oB0I2Nxx7w | feature_4 | float | None | None | None | None | 2023-07-12 12:54:24.401456+00:00 | 2023-10-14 15:42:03.557973+00:00 | 2 |
| 4 | qehni2DU75bT | feature_3 | float | None | None | None | None | 2023-07-12 12:54:24.401441+00:00 | 2023-10-14 15:42:03.431243+00:00 | 2 |
| 3 | cANjhBnEosz7 | feature_2 | float | None | None | None | None | 2023-07-12 12:54:24.401425+00:00 | 2023-10-14 15:42:03.306655+00:00 | 2 |
| 2 | RhHNXlP1jpqi | feature_1 | float | None | None | None | None | 2023-07-12 12:54:24.401408+00:00 | 2023-10-14 15:42:03.181750+00:00 | 2 |
| 1 | UwWDQLrCTdks | feature_0 | float | None | None | None | None | 2023-07-12 12:54:24.401373+00:00 | 2023-10-14 15:42:03.055457+00:00 | 2 |
308 rows × 10 columns
Let's also create a label using {class}~lamindb.ULabel, LaminDB's universal label registry.
(Later, we'll use typed labels to deal with, e.g., 100k gene identifiers.)
study = ln.ULabel(name="Candidate marker study")
study.save()
ln.ULabel.df()
| uid | name | description | reference | reference_type | created_at | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|
| id | ||||||||
| 33 | uuZlyOGk | Candidate marker study | None | None | None | 2024-03-11 16:09:13.556881+00:00 | 2024-03-11 18:39:48.336747+00:00 | 9 |
| 25 | g9VVPHHM | is_perturbation | None | None | None | 2023-10-04 08:40:48.932352+00:00 | 2024-03-11 18:33:34.534605+00:00 | 2 |
| 34 | GrFQE60W | is_study | None | None | None | 2024-03-11 16:09:14.287381+00:00 | 2024-03-11 18:33:32.310364+00:00 | 9 |
| 32 | A9uKPtTf | study0 | None | None | None | 2023-10-04 11:07:59.405582+00:00 | 2023-10-14 15:42:52.610246+00:00 | 2 |
| 31 | eNfzpckS | is_species | None | None | None | 2023-10-04 11:07:52.142208+00:00 | 2023-10-14 15:42:52.485360+00:00 | 2 |
| 30 | eF9LX1Uy | versicolor | None | None | None | 2023-10-04 11:07:43.088097+00:00 | 2023-10-14 15:42:52.360231+00:00 | 2 |
| 29 | LVS9NAYP | setosa | None | None | None | 2023-10-04 11:07:43.088075+00:00 | 2023-10-14 15:42:52.234096+00:00 | 2 |
| 28 | RAvdVFH6 | virginica | None | None | None | 2023-10-04 11:07:43.087972+00:00 | 2023-10-14 15:42:52.108691+00:00 | 2 |
| 27 | Yis4YLIB | IFNG | None | None | None | 2023-10-04 08:40:49.557375+00:00 | 2023-10-14 15:42:51.984532+00:00 | 2 |
| 26 | vmjLLqYy | DMSO | None | None | None | 2023-10-04 08:40:49.058457+00:00 | 2023-10-14 15:42:51.860325+00:00 | 2 |
| 24 | 4tuxObBQ | A35 | Donor A35 | None | None | 2023-07-26 13:13:14.744437+00:00 | 2023-10-14 15:42:51.609548+00:00 | 2 |
| 23 | iYfjnGAk | 621B | Donor 621B | None | None | 2023-07-26 13:13:14.744467+00:00 | 2023-10-14 15:42:51.485008+00:00 | 2 |
| 22 | jj7Ovwfd | A52 | Donor A52 | None | None | 2023-07-26 13:13:14.744408+00:00 | 2023-10-14 15:42:51.360348+00:00 | 2 |
| 21 | UP2UL5jL | A37 | Donor A37 | None | None | 2023-07-26 13:13:14.744379+00:00 | 2023-10-14 15:42:51.233663+00:00 | 2 |
| 20 | DvGIrrA0 | 637C | Donor 637C | None | None | 2023-07-26 13:13:14.744350+00:00 | 2023-10-14 15:42:51.108958+00:00 | 2 |
| 19 | Qjcs3YGQ | A29 | Donor A29 | None | None | 2023-07-26 13:13:14.744320+00:00 | 2023-10-14 15:42:50.984185+00:00 | 2 |
| 18 | QEhPcnIU | D503 | Donor D503 | None | None | 2023-07-26 13:13:14.744291+00:00 | 2023-10-14 15:42:50.860058+00:00 | 2 |
| 17 | C2dGiOft | 582C | Donor 582C | None | None | 2023-07-26 13:13:14.744261+00:00 | 2023-10-14 15:42:50.733881+00:00 | 2 |
| 16 | Stca6kSX | A36 | Donor A36 | None | None | 2023-07-26 13:13:14.744230+00:00 | 2023-10-14 15:42:50.608518+00:00 | 2 |
| 15 | ZME6CKuk | 640C | Donor 640C | None | None | 2023-07-26 13:13:14.744199+00:00 | 2023-10-14 15:42:50.483941+00:00 | 2 |
| 14 | U2HsiYdX | D496 | Donor D496 | None | None | 2023-07-26 13:13:14.744167+00:00 | 2023-10-14 15:42:50.359967+00:00 | 2 |
| 13 | Dj0yzIvF | A31 | Donor A31 | None | None | 2023-07-26 13:13:14.744094+00:00 | 2023-10-14 15:42:50.234212+00:00 | 2 |
| 12 | 28AmGmmD | is_donor | None | None | None | 2023-08-26 12:16:33.465282+00:00 | 2023-10-14 15:42:50.109350+00:00 | 2 |
| 11 | wf7WoZQG | S001 | primary human T cell isolated from PBMC, batch... | None | None | 2023-08-25 20:07:40.624283+00:00 | 2023-10-14 15:42:49.984548+00:00 | 2 |
| 10 | t5cjMINo | is_biosample | None | None | None | 2023-08-25 20:07:40.367235+00:00 | 2023-10-14 15:42:49.860152+00:00 | 2 |
| 9 | YYd4CsbX | EXP002 | CRISPRa Perturb-seq for characterizing molecul... | None | None | 2023-08-25 20:07:27.473277+00:00 | 2023-10-14 15:42:49.736253+00:00 | 2 |
| 8 | PmhaiMKQ | EXP001 | Genome-wide CRISPRa screen for functional regu... | None | None | 2023-08-25 20:07:24.973988+00:00 | 2023-10-14 15:42:49.611084+00:00 | 2 |
| 7 | 2NgCoWfO | is_experiment | None | None | None | 2023-08-25 20:07:13.529122+00:00 | 2023-10-14 15:42:49.485731+00:00 | 2 |
| 6 | ApoMomVX | integrated_lung | None | None | None | 2023-07-15 17:00:43.804826+00:00 | 2023-10-14 15:42:49.360376+00:00 | 3 |
| 5 | tZCTk48f | TSP14 | None | None | None | 2023-07-14 21:27:44.312320+00:00 | 2023-10-14 15:42:49.234586+00:00 | 3 |
| 4 | gk6w8qC5 | TSP2 | None | None | None | 2023-07-14 21:27:44.312301+00:00 | 2023-10-14 15:42:49.109820+00:00 | 3 |
| 3 | vfLXaHgD | TSP1 | None | None | None | 2023-07-14 21:27:44.312230+00:00 | 2023-10-14 15:42:48.985180+00:00 | 3 |
| 2 | Jel5uCyM | rxrx1 | None | None | None | 2023-07-11 15:11:32.490394+00:00 | 2023-10-14 15:42:48.861053+00:00 | 2 |
| 1 | P8HivTdn | train | None | None | None | 2023-07-05 19:22:25.674762+00:00 | 2023-10-14 15:42:48.733349+00:00 | 2 |
We can model hierachical labels like so:
is_study= ln.ULabel(name="is_study")
is_study.save()
is_study.children.add(study)
study.view_parents()

Let us do the same for perturbation labels:
perturbations = [ln.ULabel(name=label) for label in df["perturbation"].unique()]
ln.save(perturbations)
is_perturbation = ln.ULabel(name="is_perturbation")
is_perturbation.save()
is_perturbation.children.add(*perturbations)
is_perturbation.view_parents(with_children=True)

Validate & annotate¶
Now that we defined features, we can link them to the artifact:
artifact.features.add(features)
artifact.describe()
Artifact(uid='S7YB8PFudvFudzXzQMRy', suffix='.parquet', accessor='DataFrame', description='my RNA-seq', version='1', size=4122, hash='iRFVECWdNmTqDesUtIE37A', hash_type='md5', visibility=1, key_is_virtual=True, updated_at=2024-03-11 18:39:39 UTC) Provenance: 🗃️ storage: Storage(uid='D9BilDV2', root='s3://lamindata', type='s3', region='us-east-1', updated_at=2023-12-07 09:50:27 UTC, created_by_id=2) 📔 transform: Transform(uid='FPnfDtJz8qbE5zKv', name='Introduction', short_name='introduction', version='1', type='notebook', updated_at=2024-03-11 16:08:43 UTC, created_by_id=9) 👣 run: Run(uid='39aA4QIBAAYKK3OqAWNk', run_at=2024-03-11 18:39:32 UTC, transform_id=62, created_by_id=9) 👤 created_by: User(uid='FBa7SHjn', handle='falexwolf', name='Alex Wolf', updated_at=2023-10-19 18:58:28 UTC) Features: columns: FeatureSet(uid='5o3wKfvHbv7sXzFDuyH4', n=4, registry='core.Feature', hash='3M6xerbxJQKIH0SvRy9L', updated_at=2024-03-11 18:39:53 UTC, created_by_id=9) CD8A (number) CD4 (number) CD14 (number) 🔗 perturbation (2, core.ULabel): 'DMSO', 'IFNG' Labels: 🏷️ ulabels (3, core.ULabel): 'DMSO', 'IFNG', 'Candidate marker study'
Annotating an artifact with a label works like so:
artifact.labels.add(study)
artifact.describe()
Artifact(uid='S7YB8PFudvFudzXzQMRy', suffix='.parquet', accessor='DataFrame', description='my RNA-seq', version='1', size=4122, hash='iRFVECWdNmTqDesUtIE37A', hash_type='md5', visibility=1, key_is_virtual=True, updated_at=2024-03-11 18:39:39 UTC) Provenance: 🗃️ storage: Storage(uid='D9BilDV2', root='s3://lamindata', type='s3', region='us-east-1', updated_at=2023-12-07 09:50:27 UTC, created_by_id=2) 📔 transform: Transform(uid='FPnfDtJz8qbE5zKv', name='Introduction', short_name='introduction', version='1', type='notebook', updated_at=2024-03-11 16:08:43 UTC, created_by_id=9) 👣 run: Run(uid='39aA4QIBAAYKK3OqAWNk', run_at=2024-03-11 18:39:32 UTC, transform_id=62, created_by_id=9) 👤 created_by: User(uid='FBa7SHjn', handle='falexwolf', name='Alex Wolf', updated_at=2023-10-19 18:58:28 UTC) Features: columns: FeatureSet(uid='5o3wKfvHbv7sXzFDuyH4', n=4, registry='core.Feature', hash='3M6xerbxJQKIH0SvRy9L', updated_at=2024-03-11 18:39:53 UTC, created_by_id=9) CD8A (number) CD4 (number) CD14 (number) 🔗 perturbation (2, core.ULabel): 'DMSO', 'IFNG' Labels: 🏷️ ulabels (3, core.ULabel): 'DMSO', 'IFNG', 'Candidate marker study'
We can also associate labels with a feature:
features_lookup = ln.Feature.lookup()
artifact.labels.add(perturbations, feature=features_lookup.perturbation)
artifact.describe()
Artifact(uid='S7YB8PFudvFudzXzQMRy', suffix='.parquet', accessor='DataFrame', description='my RNA-seq', version='1', size=4122, hash='iRFVECWdNmTqDesUtIE37A', hash_type='md5', visibility=1, key_is_virtual=True, updated_at=2024-03-11 18:39:39 UTC) Provenance: 🗃️ storage: Storage(uid='D9BilDV2', root='s3://lamindata', type='s3', region='us-east-1', updated_at=2023-12-07 09:50:27 UTC, created_by_id=2) 📔 transform: Transform(uid='FPnfDtJz8qbE5zKv', name='Introduction', short_name='introduction', version='1', type='notebook', updated_at=2024-03-11 16:08:43 UTC, created_by_id=9) 👣 run: Run(uid='39aA4QIBAAYKK3OqAWNk', run_at=2024-03-11 18:39:32 UTC, transform_id=62, created_by_id=9) 👤 created_by: User(uid='FBa7SHjn', handle='falexwolf', name='Alex Wolf', updated_at=2023-10-19 18:58:28 UTC) Features: columns: FeatureSet(uid='5o3wKfvHbv7sXzFDuyH4', n=4, registry='core.Feature', hash='3M6xerbxJQKIH0SvRy9L', updated_at=2024-03-11 18:39:53 UTC, created_by_id=9) CD8A (number) CD4 (number) CD14 (number) 🔗 perturbation (2, core.ULabel): 'DMSO', 'IFNG' Labels: 🏷️ ulabels (3, core.ULabel): 'DMSO', 'IFNG', 'Candidate marker study'
:::{dropdown} Artifacts with context in the UI
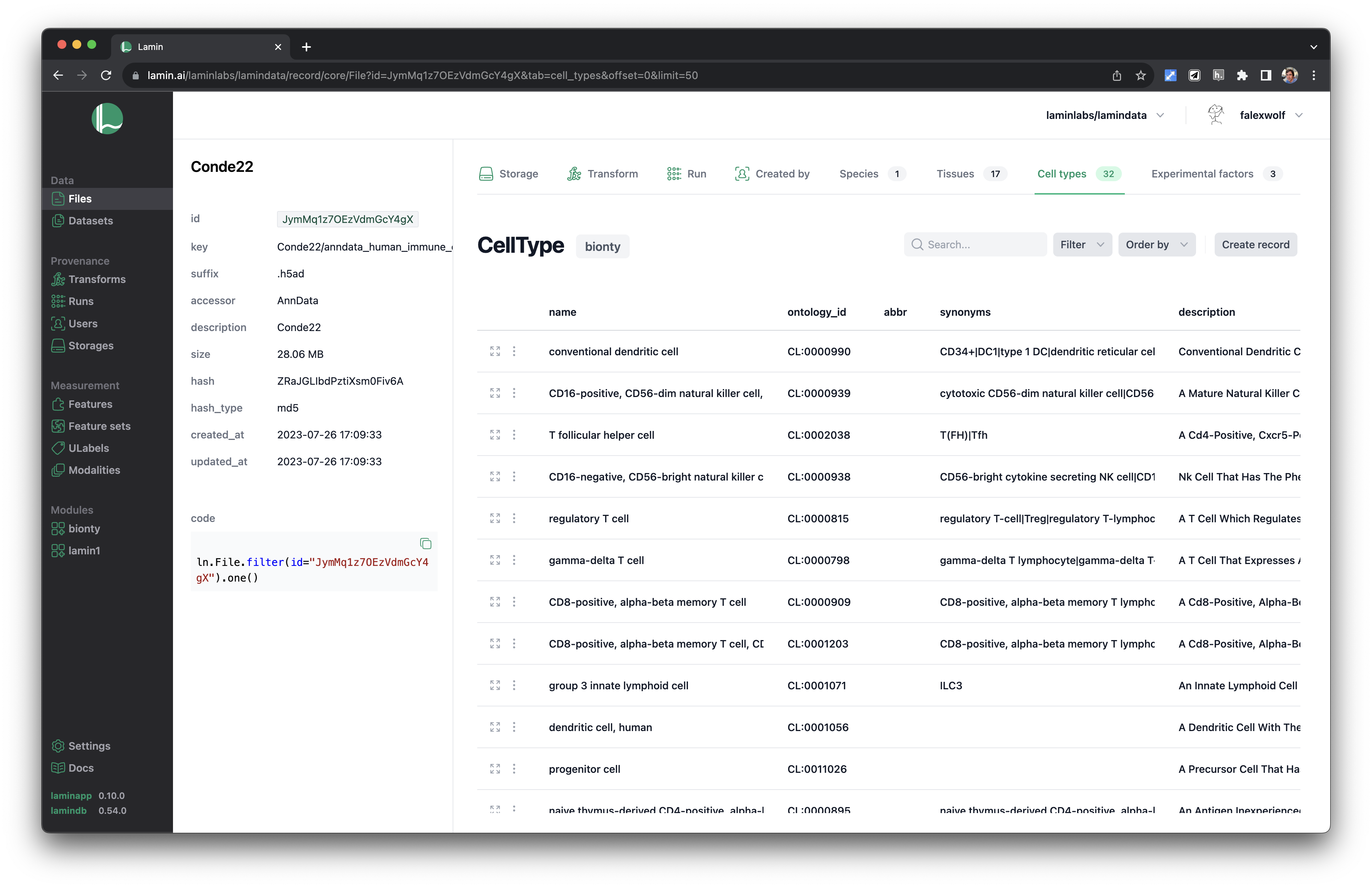
:::
Get lookup object for the entities of interest:
studies = is_study.children.lookup()
perturbations = is_perturbation.children.lookup()
ln.Artifact.filter(ulabels=studies.candidate_marker_study).filter(ulabels=perturbations.ifng).one()
Artifact(uid='S7YB8PFudvFudzXzQMRy', suffix='.parquet', accessor='DataFrame', description='my RNA-seq', version='1', size=4122, hash='iRFVECWdNmTqDesUtIE37A', hash_type='md5', visibility=1, key_is_virtual=True, updated_at=2024-03-11 18:39:39 UTC, storage_id=2, transform_id=62, run_id=77, created_by_id=9)
Delete an artifact:
artifact.delete(permanent=True)
Biological types¶
{class}~lamindb.Feature and {class}~lamindb.ULabel will get you pretty far.
But if you use an entity many times, you typically want a dedicated registry.
Let's do this with {class}~bionty.Gene and {class}~bionty.Tissue from plug-in {py:mod}bionty:
Access public ontologies¶
Import gene records from a public ontology, which we'll use to validate features:
import bionty as bt
genes = bt.Gene.from_values(df.columns, organism="human")
ln.save(genes)
bt.Gene.df()
❗ did not create Gene record for 1 non-validated symbol: 'perturbation'
| uid | symbol | stable_id | ensembl_gene_id | ncbi_gene_ids | biotype | description | synonyms | organism_id | public_source_id | created_at | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||
| 65755 | 2PcMC6Iqe3BV | TARP | None | ENSG00000289746 | None | protein_coding | TCR gamma alternate reading frame protein [Sou... | TCRGC2|TCRGV|CD3G|TCRGC1|TCRG | 2 | 3.0 | 2023-07-15 13:26:52.641339+00:00 | 2023-10-14 18:06:50.149521+00:00 | 6 |
| 65754 | 2BJhV18TyISQ | HLA-DRB4 | None | ENSG00000231021 | None | protein_coding | major histocompatibility complex, class II, DR... | HLA-DR4B | 2 | 3.0 | 2023-07-15 13:26:52.641220+00:00 | 2023-10-14 18:06:50.024899+00:00 | 6 |
| 65753 | 3OrDAjYRNL9H | HLA-DRB4 | None | ENSG00000227826 | None | protein_coding | major histocompatibility complex, class II, DR... | HLA-DR4B | 2 | 3.0 | 2023-07-15 13:26:52.641197+00:00 | 2023-10-14 18:06:49.900982+00:00 | 6 |
| 65752 | 4dPDKJKw06Ew | HLA-DRB4 | None | ENSG00000227357 | None | protein_coding | major histocompatibility complex, class II, DR... | HLA-DR4B | 2 | 3.0 | 2023-07-15 13:26:52.641175+00:00 | 2023-10-14 18:06:49.776318+00:00 | 6 |
| 65751 | FWAgOzPV9oRh | HLA-DRB3 | None | ENSG00000231679 | None | protein_coding | major histocompatibility complex, class II, DR... | HLA-DR3B | 2 | 3.0 | 2023-07-15 13:26:52.641150+00:00 | 2023-10-14 18:06:49.649087+00:00 | 6 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 5 | 3wEqb6eOTZzC | C1orf112 | None | ENSG00000000460 | None | protein_coding | chromosome 1 open reading frame 112 [Source:HG... | FLJ10706 | 2 | 3.0 | 2023-07-14 12:41:00.025449+00:00 | 2023-10-14 15:46:55.629633+00:00 | 5 |
| 4 | 5Zug63FETk4p | SCYL3 | None | ENSG00000000457 | None | protein_coding | SCY1 like pseudokinase 3 [Source:HGNC Symbol;A... | PACE1|PACE-1 | 2 | 3.0 | 2023-07-14 12:41:00.025412+00:00 | 2023-10-14 15:46:55.504719+00:00 | 5 |
| 3 | 4eC1wUNJAO2s | DPM1 | None | ENSG00000000419 | None | protein_coding | dolichyl-phosphate mannosyltransferase subunit... | CDGIE|MPDS | 2 | 3.0 | 2023-07-14 12:41:00.025375+00:00 | 2023-10-14 15:46:55.378918+00:00 | 5 |
| 2 | 2FhduD7Z97Uv | TNMD | None | ENSG00000000005 | None | protein_coding | tenomodulin [Source:HGNC Symbol;Acc:HGNC:17757] | tendin|ChM1L|TEM|myodulin|BRICD4 | 2 | 3.0 | 2023-07-14 12:41:00.025336+00:00 | 2023-10-14 15:46:55.253278+00:00 | 5 |
| 1 | 1uTi9dROoaN5 | TSPAN6 | None | ENSG00000000003 | None | protein_coding | tetraspanin 6 [Source:HGNC Symbol;Acc:HGNC:11858] | TSPAN-6|T245|TM4SF6 | 2 | 3.0 | 2023-07-14 12:41:00.025275+00:00 | 2023-10-14 15:46:55.113809+00:00 | 5 |
65755 rows × 13 columns
Validate typed features¶
To manage features of different types, let us use an AnnData object, which comes with two slots for storing data & metadata:
import anndata as ad
adata = ad.AnnData(df[["CD8A", "CD4", "CD14"]], obs=df[["perturbation"]])
Create an artifact & validate features using the symbol field of Gene:
artifact = ln.Artifact.from_anndata(adata, description="my RNA-seq")
artifact.save()
... storing 'perturbation' as categorical
... uploading Tru6AqMMSDsFIAMvuqNx.h5ad: 100.0%
Annotate with typed labels¶
Search the public tissue ontology from the bionty store:
bt.Tissue.public().search("umbilical blood").head(2)
| ontology_id | definition | synonyms | parents | __ratio__ | |
|---|---|---|---|---|---|
| name | |||||
| umbilical cord blood | UBERON:0012168 | Blood That Remains In The Placenta And In The ... | None | [UBERON:0013755] | 85.714286 |
| umbilical cord | UBERON:0002331 | The Connecting Cord From The Developing Embryo... | chorda umbilicalis|funiculus umbilicalis | [UBERON:0000478] | 82.758621 |
Define a tissue label:
tissue = bt.Tissue.from_public(name="umbilical cord blood")
tissue.save()
tissue.view_parents(distance=2)

Annotate the artifact:
artifact.features.add_from_anndata(var_field=bt.Gene.symbol, organism="human")
artifact.labels.add(study)
artifact.labels.add(adata.obs.perturbation, feature=features_lookup.perturbation)
artifact.labels.add(tissue)
artifact.describe()
Artifact(uid='Tru6AqMMSDsFIAMvuqNx', suffix='.h5ad', accessor='AnnData', description='my RNA-seq', size=19240, hash='ohAeiVMJZOrc3bFTKmankw', hash_type='md5', visibility=1, key_is_virtual=True, updated_at=2024-03-11 18:40:48 UTC) Provenance: 🗃️ storage: Storage(uid='D9BilDV2', root='s3://lamindata', type='s3', region='us-east-1', updated_at=2023-12-07 09:50:27 UTC, created_by_id=2) 📔 transform: Transform(uid='FPnfDtJz8qbE5zKv', name='Introduction', short_name='introduction', version='1', type='notebook', updated_at=2024-03-11 16:08:43 UTC, created_by_id=9) 👣 run: Run(uid='39aA4QIBAAYKK3OqAWNk', run_at=2024-03-11 18:39:32 UTC, transform_id=62, created_by_id=9) 👤 created_by: User(uid='FBa7SHjn', handle='falexwolf', name='Alex Wolf', updated_at=2023-10-19 18:58:28 UTC) Features: var: FeatureSet(uid='4BCNJcOI7wLozkA5y3X8', n=3, type='number', registry='bionty.Gene', hash='f2UVeHefaZxXFjmUwo9O', updated_at=2024-03-11 16:09:48 UTC, created_by_id=9) 'CD4', 'CD8A', 'CD14' obs: FeatureSet(uid='gZs5VXAT6RU2H8jaxPtn', n=1, registry='core.Feature', hash='ymtT1QE9yB6Rp41F9vw0', updated_at=2023-10-19 20:57:28 UTC, created_by_id=2) 🔗 perturbation (2, core.ULabel): 'DMSO', 'IFNG' Labels: 🏷️ tissues (1, bionty.Tissue): 'umbilical cord blood' 🏷️ ulabels (3, core.ULabel): 'DMSO', 'IFNG', 'Candidate marker study'
Query for genes & the linked artifacts:
genes = bt.Gene.filter(organism__name="human").lookup()
# all gene sets measuring CD8A
genesets_with_cd8a = ln.FeatureSet.filter(genes=genes.cd8a).all()
# all artifacts measuring CD8A
ln.Artifact.filter(feature_sets__in=genesets_with_cd8a).df()
| uid | storage_id | key | suffix | accessor | description | version | size | hash | hash_type | n_objects | n_observations | transform_id | run_id | visibility | key_is_virtual | created_at | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||
| 17 | DDFxKYXoNMmHzQAHqpx0 | 2 | None | .h5mu | None | Papalexi 2021 | None | 617665133 | fCEddup3HMzqYT5fArpd2i | sha1-fl | None | None | 22 | 16 | 1 | False | 2023-07-14 19:58:52.403089+00:00 | 2023-10-14 15:40:52.058720+00:00 | 9 |
| 446 | xJkeL0OxEFIpvGWKdpne | 2 | scrna/conde22.h5ad | .h5ad | AnnData | Human immune cells from Conde22 | None | 57612943 | 9sXda5E7BYiVoDOQkTC0KB | sha1-fl | None | None | 42 | 8 | 1 | True | 2023-11-08 09:56:02.106561+00:00 | 2024-01-08 12:34:54.789845+00:00 | 2 |
| 383 | Ff4XZtm6Lko0Fz3ysIEl | 2 | None | .h5ad | AnnData | 10x reference adata | None | 660792 | TRHLp5EVjDGdv7DufMAnmg | md5 | None | None | 16 | 33 | 1 | False | 2023-10-04 13:19:07.817861+00:00 | 2023-10-14 15:41:39.068835+00:00 | 2 |
| 453 | ckBmWq8Bqzmej574ogH5 | 2 | None | .h5ad | AnnData | 10x reference adata | None | 857752 | R4CCdaE26v2hikWjBVJO9w | md5 | None | None | 55 | 55 | 1 | True | 2024-01-03 00:41:58.632361+00:00 | 2024-01-03 00:41:58.632367+00:00 | 9 |
| 468 | Tru6AqMMSDsFIAMvuqNx | 2 | None | .h5ad | AnnData | my RNA-seq | None | 19240 | ohAeiVMJZOrc3bFTKmankw | md5 | None | None | 62 | 77 | 1 | True | 2024-03-11 18:40:39.584218+00:00 | 2024-03-11 18:40:48.608663+00:00 | 9 |
# create an ontology-coupled cell type record and save it
bt.CellType.from_public(name="neuron").save()
# create a record to track a new cell state
new_cell_state = bt.CellType(name="my neuron cell state", description="explains X")
new_cell_state.save()
# express that it's a neuron state
cell_types = bt.CellType.lookup()
new_cell_state.parents.add(cell_types.neuron)
# view ontological hierarchy
new_cell_state.view_parents(distance=2)

Collections of artifacts¶
Assume we now run a pipeline in which we access a new batch of data:
transform = ln.Transform(name="Cell Ranger", type="pipeline", version="1")
ln.track(transform)
💡 saved: Transform(uid='IUU9JQ50xyjLtmKZ', name='Cell Ranger', version='1', type='pipeline', updated_at=2024-03-11 18:41:08 UTC, created_by_id=9) 💡 saved: Run(uid='rpacdPZ1XeulYz0gBMHv', run_at=2024-03-11 18:41:08 UTC, transform_id=63, created_by_id=9)
Access a new batch of data:
df = pd.DataFrame(
{
"CD8A": [2, 3, 3],
"CD4": [3, 4, 5],
"CD38": [4, 2, 3],
"perturbation": ["DMSO", "IFNG", "IFNG"]
},
index=["observation4", "observation5", "observation6"],
)
adata = ad.AnnData(df[["CD8A", "CD4", "CD38"]], obs=df[["perturbation"]])
Because gene "CD38" is not yet registered, it doesn't yet validate:
bt.Gene.validate(adata.var_names, field=bt.Gene.symbol, organism="human");
Let's add it to the Gene registry and link to the artifact - now all features validate:
bt.Gene.from_public(symbol="CD38", organism="human").save()
artifact2 = ln.Artifact.from_anndata(
adata, description="my RNA-seq batch 2"
)
artifact2.save()
artifact2.features.add_from_anndata(var_field=bt.Gene.symbol, organism="human")
artifact2.describe()
... storing 'perturbation' as categorical
... uploading WqbgfPlHdrjKUOBP8Gzb.h5ad: 100.0% Artifact(uid='WqbgfPlHdrjKUOBP8Gzb', suffix='.h5ad', accessor='AnnData', description='my RNA-seq batch 2', size=19240, hash='L37UPl4IUH20HkIRzvlRMw', hash_type='md5', visibility=1, key_is_virtual=True, updated_at=2024-03-11 18:41:20 UTC) Provenance: 🗃️ storage: Storage(uid='D9BilDV2', root='s3://lamindata', type='s3', region='us-east-1', updated_at=2023-12-07 09:50:27 UTC, created_by_id=2) 🧩 transform: Transform(uid='IUU9JQ50xyjLtmKZ', name='Cell Ranger', version='1', type='pipeline', updated_at=2024-03-11 18:41:08 UTC, created_by_id=9) 👣 run: Run(uid='rpacdPZ1XeulYz0gBMHv', run_at=2024-03-11 18:41:08 UTC, transform_id=63, created_by_id=9) 👤 created_by: User(uid='FBa7SHjn', handle='falexwolf', name='Alex Wolf', updated_at=2023-10-19 18:58:28 UTC) Features: var: FeatureSet(uid='iQaFYclhBHotmbHXqr9V', n=3, type='number', registry='bionty.Gene', hash='QW2rHuIo5-eGNZbRxHMD', updated_at=2024-03-11 18:41:20 UTC, created_by_id=9) 'CD38', 'CD4', 'CD8A' obs: FeatureSet(uid='gZs5VXAT6RU2H8jaxPtn', n=1, registry='core.Feature', hash='ymtT1QE9yB6Rp41F9vw0', updated_at=2023-10-19 20:57:28 UTC, created_by_id=2) 🔗 perturbation (0, core.ULabel):
Create a collection using {class}~lamindb.Collection:
collection = ln.Collection([artifact, artifact2], name="my RNA-seq collection", version="1")
collection.save()
collection.describe()
collection.view_lineage()
Collection(uid='F2VLEUmTdWruY4gKpKOR', name='my RNA-seq collection', version='1', hash='5g0aLY_lBSTkIYYUTycd', visibility=1, updated_at=2024-03-11 18:41:29 UTC) Provenance: 🧩 transform: Transform(uid='IUU9JQ50xyjLtmKZ', name='Cell Ranger', version='1', type='pipeline', updated_at=2024-03-11 18:41:08 UTC, created_by_id=9) 👣 run: Run(uid='rpacdPZ1XeulYz0gBMHv', run_at=2024-03-11 18:41:08 UTC, transform_id=63, created_by_id=9) 👤 created_by: User(uid='FBa7SHjn', handle='falexwolf', name='Alex Wolf', updated_at=2023-10-19 18:58:28 UTC) Features: var: FeatureSet(uid='YsoDRKWF1QjRYLYjCA4i', n=4, type='number', registry='bionty.Gene', hash='yJG4sMJqJjN0dRBkDgYZ', updated_at=2024-03-11 18:41:28 UTC, created_by_id=9) 'CD38', 'CD4', 'CD14', 'CD8A' obs: FeatureSet(uid='gZs5VXAT6RU2H8jaxPtn', n=1, registry='core.Feature', hash='ymtT1QE9yB6Rp41F9vw0', updated_at=2023-10-19 20:57:28 UTC, created_by_id=2) 🔗 perturbation (0, core.ULabel):

If it's small enough, you can load the entire collection into memory as if it was one:
collection.load()
AnnData object with n_obs × n_vars = 6 × 4
obs: 'perturbation', 'artifact_uid'
Iterate over its artifacts:
collection.artifacts.df()
| uid | storage_id | key | suffix | accessor | description | version | size | hash | hash_type | n_objects | n_observations | transform_id | run_id | visibility | key_is_virtual | created_at | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||
| 468 | Tru6AqMMSDsFIAMvuqNx | 2 | None | .h5ad | AnnData | my RNA-seq | None | 19240 | ohAeiVMJZOrc3bFTKmankw | md5 | None | None | 62 | 77 | 1 | True | 2024-03-11 18:40:39.584218+00:00 | 2024-03-11 18:40:48.608663+00:00 | 9 |
| 469 | WqbgfPlHdrjKUOBP8Gzb | 2 | None | .h5ad | AnnData | my RNA-seq batch 2 | None | 19240 | L37UPl4IUH20HkIRzvlRMw | md5 | None | None | 63 | 78 | 1 | True | 2024-03-11 18:41:14.733545+00:00 | 2024-03-11 18:41:20.990693+00:00 | 9 |
Save notebooks & scripts¶
If you run lamin save <notebook_or_script_path>, you save execution report, source code and compute environment to your default storage location.
See an example for this introductory notebook here.
:::{dropdown} Show me a screenshot
:::
If you want to download a notebook or script, call:
lamin stage https://lamin.ai/laminlabs/lamindata/transform/FPnfDtJz8qbEz8
Machine learning models¶
Using {class}~lamindb.core.MappedCollection you can train machine learning models on large collections of artifacts:
from torch.utils.data import DataLoader, WeightedRandomSampler
dataset = collection.mapped(label_keys=["perturbation"])
sampler = WeightedRandomSampler(
weights=dataset.get_label_weights("perturbation"), num_samples=len(dataset)
)
dl = DataLoader(dataset, batch_size=2, sampler=sampler)
for batch in dl:
pass
Data lineage¶
View the sequence of data transformations ({class}~lamindb.Transform) in a project (from here, based on Schmidt et al., 2022):
transform.view_parents()

Or, the generating flow of an artifact:
artifact.view_lineage()

Both figures are based on mere calls to ln.track() in notebooks, pipelines & app.
Mesh of databases¶
LaminDB is a distributed system like git. Similar to cloning a repository, collaborators can load your instance on the command-line using:
lamin load myhandle/myinstance
Custom schemas¶
LaminDB can be customized & extended with schema & app plug-ins building on the Django ecosystem. Examples are
- bionty: Registries for basic biological entities, coupled to public ontologies.
- wetlab: Exemplary custom schema to manage samples, treatments, etc.
If you'd like to create your own schema or app:
- Create a git repository with registries similar to wetlab
- Create & deploy migrations via
lamin migrate createandlamin migrate deploy
It's fastest if we do this for you based on our templates within an enterprise plan.
Design¶
Why?¶
See this blog post.
Schema & API¶
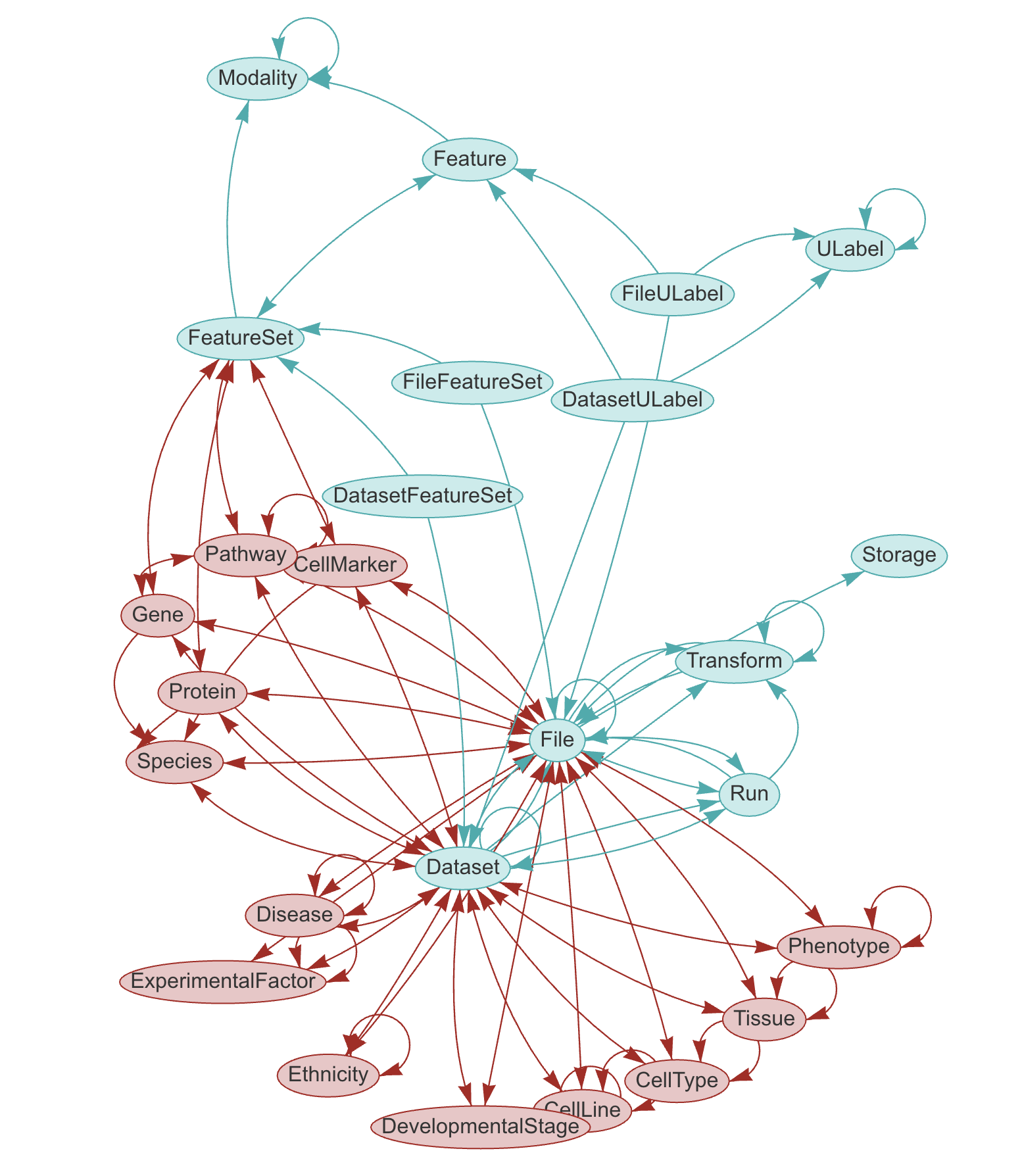
LaminDB provides a SQL schema for common entities: {class}~lamindb.Artifact, {class}~lamindb.Collection, {class}~lamindb.Transform, {class}~lamindb.Feature, {class}~lamindb.ULabel etc. - see the API reference or the source code.
The core schema is extendable through plug ins (see blue vs. red entities in graphic), e.g., with basic biological ({class}~bionty.Gene, {class}~bionty.Protein, {class}~bionty.CellLine, etc.) & operational entities (Biosample, Techsample, Treatment, etc.).
{dropdown}
Data models are defined in Python using the Django ORM. Django translates them to SQL tables.
[Django](https://github.com/django/django) is one of the most-used & highly-starred projects on GitHub (~1M dependents, ~73k stars) and has been robustly maintained for 15 years.
In the first year, LaminDB used SQLModel/SQLAlchemy -- we might bring back compatibility.
On top of the schema, LaminDB is a Python API that abstracts over storage & database access, data transformations, and (biological) ontologies.
The code for this is open-source & accessible through the dependencies & repositories listed below.
Dependencies¶
- Data is stored in a platform-independent way:
- location → local, on AWS S3 or GCP Storage, accessed through
fsspec - format → blob-like artifacts or queryable formats like parquet, zarr, HDF5, TileDB, ...
- location → local, on AWS S3 or GCP Storage, accessed through
- Metadata is stored in SQL: current backends are SQLite (small teams) and Postgres (any team size).
- Django ORM for schema management & metadata queries.
- Biological knowledge sources & ontologies: see Bionty.
For more details, see the pyproject.toml artifact in lamindb & the linked repositories below.
Repositories¶
LaminDB and its plug-ins consist in open-source Python libraries & publicly hosted metadata assets:
- lamindb: Core API, which builds on the core schema.
- bionty: Registries for basic biological entities, coupled to public ontologies.
- wetlab: Exemplary custom schema to manage samples, treatments, etc.
- lamindb-setup: Setup & configure LaminDB, client for LaminHub.
- lamin-cli: CLI for
lamindbandlamindb-setup. - nbproject: Metadata parser for Jupyter notebooks.
- lamin-utils: Generic utilities, e.g., a logger.
- readfcs: FCS artifact reader.
LaminHub is not open-sourced, and neither are plug-ins that model lab operations.
Assumptions & principles¶

- Data is generated in batches by instruments that process physical samples.
- Batches are transformed into more useful representations
- Semantics of high-level embeddings ("inflammatory", "lipophile") are anchored in experimental metadata and knowledge (ontologies)
- Experimental metadata is another ontology type
- Experiments measure features ({class}
~lamindb.Feature, {class}~bionty.CellMarker, ...) - Samples are annotated by labels ({class}
~lamindb.ULabel, {class}~bionty.CellLine, ...) - Learning and data warehousing both iterate transformations (see graphic, {class}
~lamindb.Transform) - Basic biological entities should have the same meaning to anyone and across any data platform
Influences¶
LaminDB was influenced by many other projects, see {doc}docs:influences.
Notebooks¶
- Find all tutorial & guide notebooks here and use cases here.
- You can run these notebooks in hosted versions of JupyterLab, e.g., Saturn Cloud, Google Vertex AI, Google Colab, and others.
# clean up test instance
!lamin delete --force lamin-intro
!rm -r lamin-intro
💡 deleting instance falexwolf/lamin-intro ❗ manually delete your stored data: /Users/falexwolf/repos/laminapp-ui/rest-app/sub/lamindb/docs/lamin-intro